本文最后更新于 176 天前,其中的信息可能已经有所发展或是发生改变。
分析前准备
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -ur compact
#选0,去除周期性边界条件 (PBC, Periodic Boundary Conditions) 的影响,并生成一个更直观的轨迹文件,方便后续分析与可视化RMSD
gmx rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg #选1和4RMSF
gmx rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 95000 -e 100000 -res -o rmsf_95_100.xvg #选4氢键
gmx hbond -s md_0_1.tpr -f md_0_1.xtc -num hbond.xvg #选1和13回旋半径
gmx trjconv -s md_0_1.tpr -f md_0_1.xtc -n index.ndx -b 19999 -e 20000 -o md_20_ligand.pdb -pbc mol -center receptor and ligand #截取做动态图自由能形貌图
在计算 RMSD 和回旋半径(Rg)时,我们已经获得了 rmsd_ps.xvg 和 gyrate.xvg 这两个文件。通过命令 “python . /pc_combine.py rmsd_ps.xvg gyrate.xvg output.xvg” 可以将这两个文件合并成 output.xvg。pc_combine.py 脚本如下:
import os
import sys
def main():
file1, file2, outfile = "", "", ""
cmd = sys.argv[1:]
if len(cmd) == 0 :
print(" Usage : pc_combine.py pc1 pc2 output-file ")
exit()
elif len(cmd) == 3:
file1 = cmd[0]
file2 = cmd[1]
outfile = cmd[2]
else:
print("wrong input, check it")
exit()
# deal with file
with open(file1, 'r') as fo:
content1 = fo.read()
with open(file2, 'r') as fo:
content2 = fo.read()
lines1 = content1.strip().strip("\n").split("\n")
lines2 = content2.strip().strip("\n").split("\n")
data1 = [li.strip().split() for li in lines1 if li[0] != '@' and li[0] != '&' and li[0] != '#']
data2 = [li.strip().split() for li in lines2 if li[0] != '@' and li[0] != '&' and li[0] != '#']
# print(data1)
# print(data2)
output = ""
if len(data1) != len(data2):
print("wrong length of data1 and data2, check it ")
exit()
for i in range(len(data1)):
if float(data1[i][0]) == float(data2[i][0]):
output += "{:16} {:16} {:16} \n".format(data1[i][0], data1[i][1], data2[i][1])
with open(outfile, "w") as fo:
fo.write(output)
print("done")
if __name__ == "__main__":
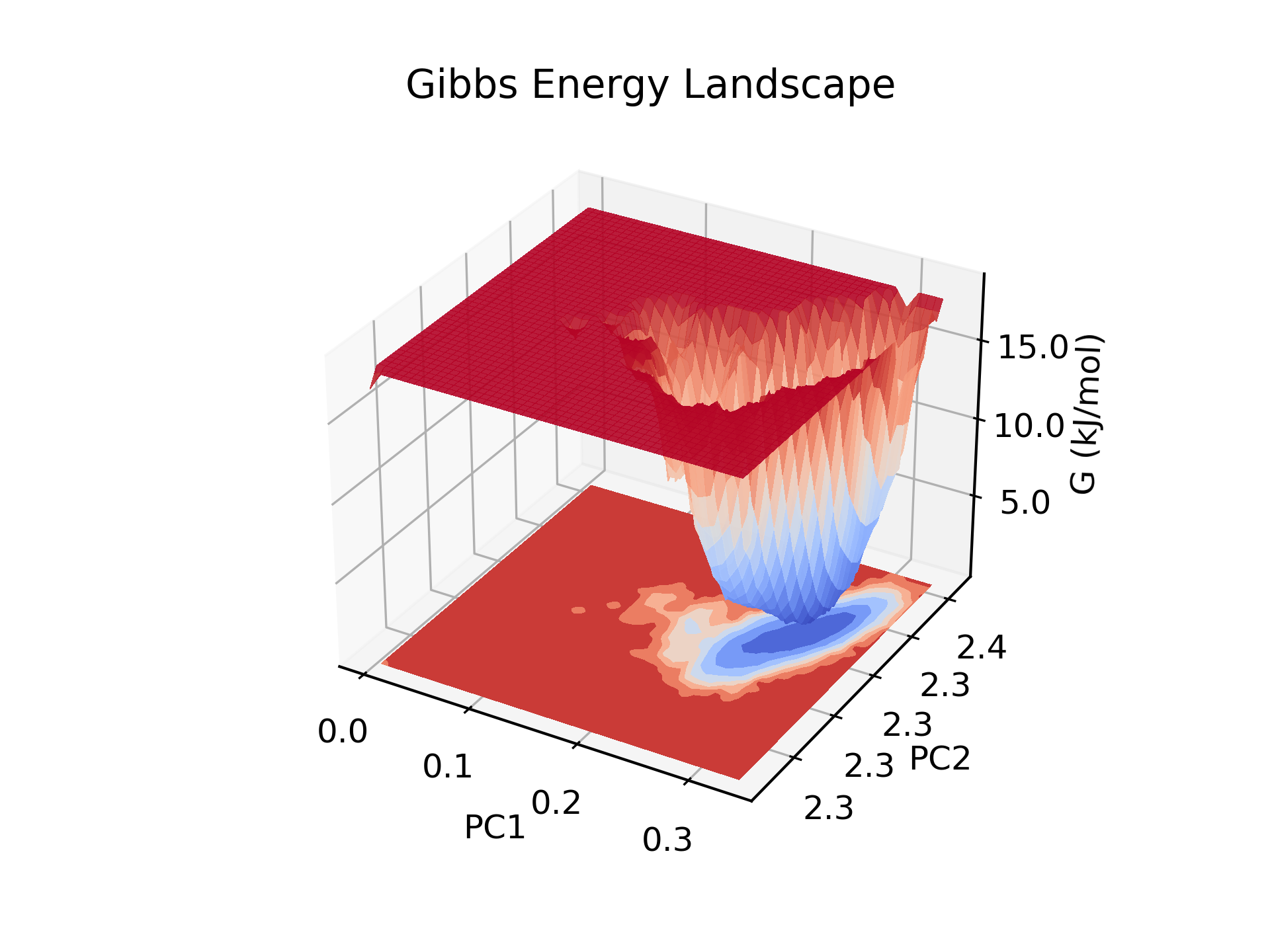
main()合并后,通过命令 “gmx sham -tsham 310 -nlevels 100 -f output.xvg -ls gibbs.xpm -g gibbs.log” 生成 gibbs.xpm 文件。再使用命令 “dito xpm_show -f gibbs.xpm -ip -o gibbs.pdf” 和 “dito xpm_show -f gibbs.xpm -3d -ip -o gibbs_3d.pdf” 即可获得 2D 和 3D 的自由能形貌图。
gmx sham -tsham 310 -nlevels 100 -f output.xvg -ls gibbs.xpm -g gibbs.log #生成gibbs.xpm
dito xpm_show -f gibbs.xpm -ip - o gibbs.pdf #2D自由能形貌图
dito xpm_show -f gibbs.xpm -3d -ip -o gibbs_3d.pdf #3D自由能形貌图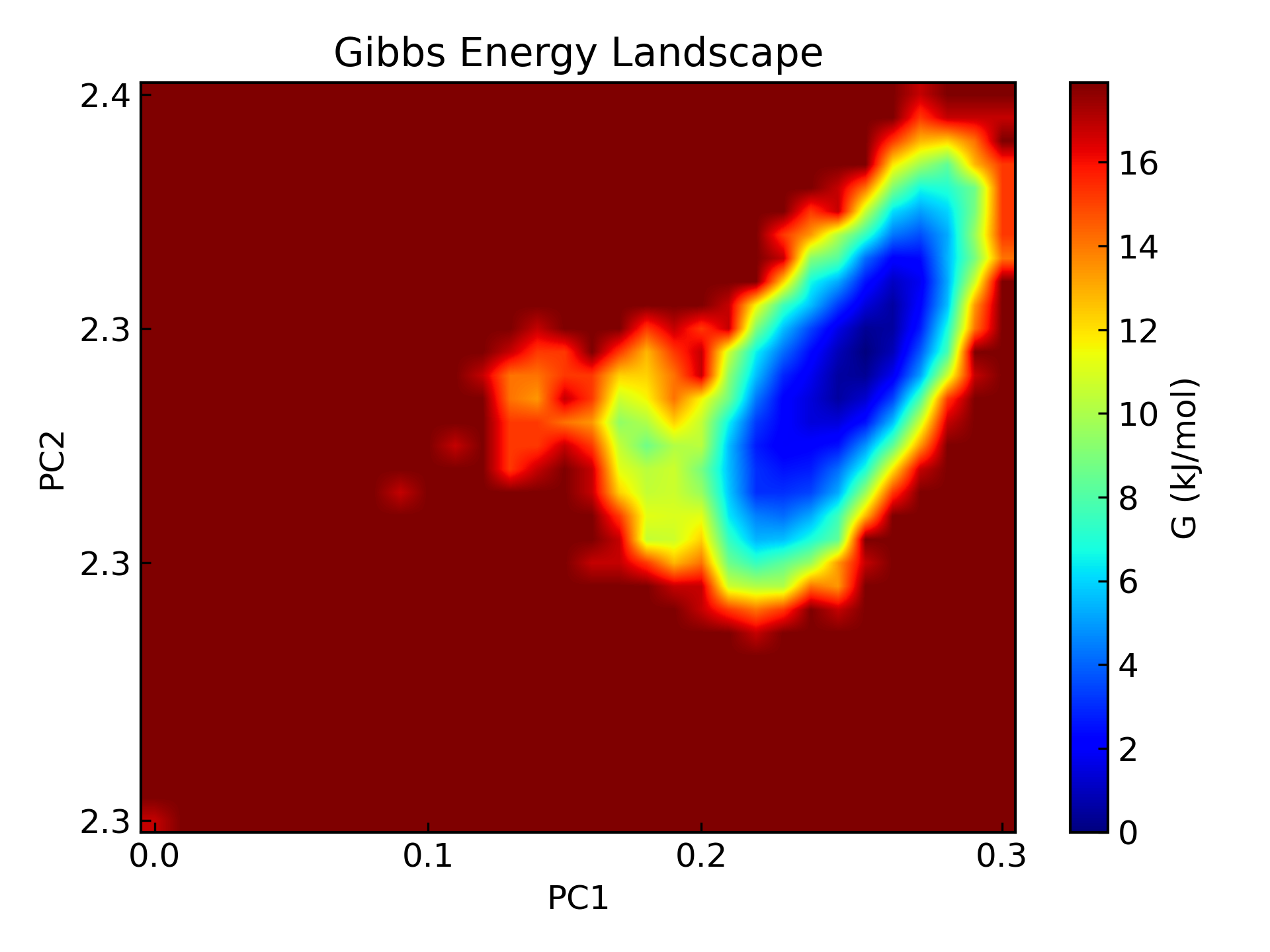
动态交叉关联矩阵(DCCM)
在做分子对接时,我们经常会分析小分子和蛋白残基之间的相互作用,而在分子动力学模拟时分析时欠缺了一些。通常只通过RMSD、RMSF、Rg和氢键这些指标去评价蛋白-小分子复合物体系的稳定性,而很少通过分子动力学模拟去分析小分子激动或抑制蛋白的分子机制。DCCM对于关键残基的识别、配体结合、突变效应和长距离相互作用的分析非常重要,具有分析的价值。通过下面的python代码可以直接出图,输入文件只需要模拟的结果文件“md.gro”和“md.xtc”。
import numpy as np
import matplotlib.pyplot as plt
import matplotlib
import MDAnalysis as mda
from MDAnalysis.analysis import align
import sys
import os
import warnings
warnings.filterwarnings('ignore')
def main(gro_file, xtc_file):
"""主函数,优化处理流程"""
# 1. 创建Universe对象
u = mda.Universe(gro_file, xtc_file)
# 2. 选择CA原子
ca_atoms = u.select_atoms("protein and name CA")
n_ca = len(ca_atoms)
n_frames = len(u.trajectory)
if n_ca == 0:
print("错误:没有选择到CA原子。请检查gro文件格式和选择条件。")
print("尝试选择所有原子作为替代方案...")
ca_atoms = u.select_atoms("all")
n_ca = len(ca_atoms)
print(f"处理轨迹:{n_frames} 帧, {n_ca} 个原子")
# 3. 获取参考结构(第一帧)
u.trajectory[0]
reference = ca_atoms.positions.copy()
# 4. 流式处理轨迹
all_coords = np.zeros((n_frames, n_ca, 3))
# 5. 使用MDAnalysis内置对齐功能(更高效)
alignment = align.AlignTraj(u, u, select="protein and name CA", in_memory=False)
alignment.run()
# 6. 收集对齐后的坐标
for i, ts in enumerate(u.trajectory):
if i % 100 == 0:
print(f"处理帧 {i+1}/{n_frames}")
all_coords[i] = ca_atoms.positions.copy()
# 7. 计算DCCM
print("计算DCCM...")
# 计算平均结构
mean_coords = np.mean(all_coords, axis=0)
# 计算偏差
deviations = all_coords - mean_coords[np.newaxis, :, :]
# 预分配内存
cij = np.zeros((n_ca, n_ca))
diag = np.zeros(n_ca)
# 计算自相关
for i in range(n_ca):
diag[i] = np.mean(np.sum(deviations[:, i, :] * deviations[:, i, :], axis=1))
# 计算互相关
for i in range(n_ca):
for j in range(i, n_ca):
dot_products = np.sum(deviations[:, i, :] * deviations[:, j, :], axis=1)
corr = np.mean(dot_products) / np.sqrt(diag[i] * diag[j])
cij[i, j] = corr
cij[j, i] = corr # 对称矩阵
# 8. 绘图
print("绘制DCCM图...")
plt.figure(figsize=(14, 12))
cmap1 = matplotlib.colors.ListedColormap(['#ff80ff', "#ffabff", "#ffd4ff", "#fffcff", "#d4ffff", "#a8ffff", "#80ffff"])
norm = matplotlib.colors.BoundaryNorm([-1.0, -0.75, -0.5, -0.25, 0.25, 0.5, 0.75, 1.0], cmap1.N)
plt.contourf(cij, cmap=cmap1, levels=[-1, -0.75, -0.5, -0.25, 0.25, 0.5, 0.75, 1], norm=norm)
plt.colorbar()
plt.contour(cij, colors="grey", levels=[-1, -0.75, -0.5, -0.25, 0.25, 0.5, 0.75, 1], linewidths=0.8, linestyles="solid")
ax = plt.gca()
font1 = {'family': 'Arial', 'weight': 'normal', 'size': 24}
ax.set_xlabel("残基编号", font1, labelpad=15.0)
ax.set_ylabel("残基编号", font1, labelpad=15.0)
# 设置刻度间隔(根据原子数量调整)
max_ticks = min(20, n_ca) # 最多20个刻度
tick_interval = max(1, n_ca // max_ticks)
ax.xaxis.set_major_locator(matplotlib.ticker.MultipleLocator(tick_interval))
ax.yaxis.set_major_locator(matplotlib.ticker.MultipleLocator(tick_interval))
ax.set_title("残基交叉相关图", fontweight="bold", fontsize=26, pad=20)
# 添加残基编号标签(仅当原子数小于100时)
if n_ca < 100:
residue_numbers = [atom.residue.resnum for atom in ca_atoms]
ax.set_xticks(range(n_ca))
ax.set_yticks(range(n_ca))
ax.set_xticklabels(residue_numbers, rotation=90, fontsize=8)
ax.set_yticklabels(residue_numbers, fontsize=8)
plt.savefig("DCCM.pdf", dpi=300, bbox_inches='tight')
plt.savefig("DCCM.png", dpi=300, bbox_inches='tight')
print("DCCM图已保存为 DCCM.pdf")
# 9. 可选:保存相关矩阵数据
np.save("dccm_matrix.npy", cij)
print("相关矩阵已保存为 dccm_matrix.npy")
if __name__ == "__main__":
if len(sys.argv) < 3:
print("使用方法: python dccm.py <gro文件> <xtc文件>")
print("示例: python dccm.py system.gro trajectory.xtc")
sys.exit(1)
try:
main(sys.argv[1], sys.argv[2])
except Exception as e:
print(f"发生错误: {str(e)}")
print("请确保已安装MDAnalysis: pip install mdanalysis")目前代码有些小问题,横纵标签可能无法正确输出,需要作图时手动添加。
