REMD介绍
简介
副本交换分子动力学模拟(Replica Exchange Molecular Dynamics, REMD),也称为并行回火(Parallel Tempering),是一种增强抽样分子模拟技术。它通过同时运行多个在不同温度(或其他热力学参数)下的模拟副本,并允许这些副本根据一定的规则交换构型,从而有效地克服能垒,加速系统的构型空间采样。目前少见用于药学领域。
核心思想
REMD的核心思想是通过在多个温度下并行运行模拟,并允许相邻温度的模拟副本之间交换构型。高温下的模拟可以跨越能垒,探索更广阔的构型空间;而低温下的模拟则可以精细地探索局部能阱。通过交换,低温副本可以获得高温副本探索到的构型,从而避免陷入局部极小值。简单的说,相较于普通的分子动力学模拟,REMD能消耗更少的计算资源探索出结合能更低的构象。
工作原理
1.多个副本:模拟同时在多个温度(T0, T1, …, Tn)下进行,其中T0是目标温度,其他温度通常更高。
2.交换尝试:定期尝试交换相邻温度副本的构型。交换概率由Metropolis准则决定:交换接受概率基于两个副本的能量和温度差计算。
3.增强采样:高温副本快速探索构型空间,低温副本提供低温下的精确采样。通过交换,所有副本都能更有效地采样。
详细操作
构建复合物体系并进行能量最小化
和常规的分子动力学模拟一样,做到NVT平衡前一步。
gmx editconf -f out_ligand1_addH.pdb -o LIG.gro #将蛋白转换成gro格式文件
gmx pdb2gmx -f 7alv-8etr.pdb -o receptor_processed.gro -water spce 力场选54a7 #使用54a7立场处理蛋白
# 将上一步生成的receptor.gro文件与配体的LIG.gro文件合并到complex.gro
# 在Topol.top文件的底部加入下列两行
; Include ligand topology
#include "LIG.itp"
#在Topol.top文件的最后一行加入下面这行
LIG 1
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0 # 将复合物置于周期性水盒子中
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr -maxwarn 200
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -np 5 # 电荷平衡
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr -maxwarn 200 # 能量最小化
gmx mdrun -v -deffnm em -nb gpu
gmx genrestr -f LIG.gro -o posre_LIG.itp -fc 1000 1000 1000 #约束配体
# 在Topol.top文件的; Include ligand topology #include "LIG.itp" 后面加入下面四行
; Ligand position restraints
#ifdef POSRES
#include "posre_LIG.itp"
#endif 计算副本温度
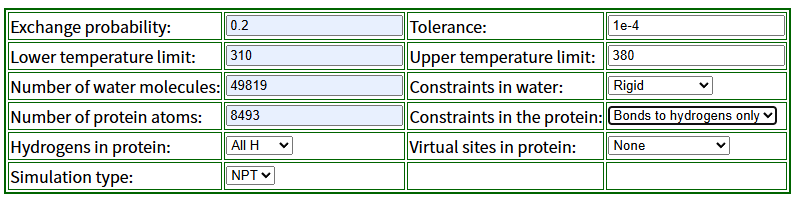
通过网站 http://virtualchemistry.org/remd-temperature-generator/ 计算副本温度
执行NVT、NPT平衡
执行下面的脚本,执行前先获取当前目录的所有权和读写权限,同时给脚本加上权限
sudo chown -R $USER:$USER .
sudo chmod -R u+rwx .
sudo chmod 777 step1.sh现在,执行下面的脚本吧! 别忘了将温度替换成上面计算的副本温度。
#!/bin/bash
T="310.00, 310.79, 311.59, 312.38, 313.18, 313.98, 314.78, 315.58, 316.39, 317.19, 318.00, 318.81, 319.62, 320.44, 321.25, 322.07, 322.89, 323.70, 324.53, 325.35, 326.18, 327.00, 327.83, 328.66, 329.49, 330.33, 331.16, 332.00, 332.84, 333.68, 334.52, 335.37, 336.21, 337.06, 337.91, 338.76, 339.61, 340.47, 341.32, 342.18, 343.04, 343.90, 344.77, 345.63, 346.50, 347.37, 348.24, 349.12, 349.99, 350.87, 351.75, 352.63, 353.51, 354.39, 355.28, 356.17, 357.05, 357.95, 358.84, 359.73, 360.63, 361.53, 362.43, 363.33, 364.24, 365.16, 366.07, 366.98, 367.89, 368.80, 369.72, 370.64, 371.55, 372.48, 373.40, 374.33, 375.25, 376.18, 377.11, 378.05, 378.98, 379.92, 380.00"
arr_T=(${T//,/ }) #温度数据转为数组
len=${#arr_T[@]} #统计数组元素个数
rm -r ./step1/equ* > /dev/null 2>&1 #删除step1路径下的残存文件夹
for ((i=0; i<=$len-1; i++))
do
mkdir ./step1/equ_${i}
cp ./step1/mdp_file/* ./step1/equ_${i}/.
sed -i 's/TEMP/'${arr_T[$i]}'/g' ./step1/equ_${i}/*.mdp
#预平衡
gmx_mpi grompp -f ./step1/equ_${i}/PR.mdp -c em.gro -r em.gro -p topol.top -o ./step1/equ_${i}/2PR.tpr -maxwarn 200
gmx_mpi mdrun -deffnm ./step1/equ_${i}/2PR -v
#NVT
gmx_mpi grompp -f ./step1/equ_${i}/NVT.mdp -c ./step1/equ_${i}/2PR.gro -p topol.top -o ./step1/equ_${i}/3NVT.tpr -maxwarn 200
gmx_mpi mdrun -deffnm ./step1/equ_${i}/3NVT -v
#NPT
gmx_mpi grompp -f ./step1/equ_${i}/NPT.mdp -c ./step1/equ_${i}/3NVT.gro -p topol.top -o ./step1/equ_${i}/4NPT.tpr -maxwarn 200
gmx_mpi mdrun -deffnm ./step1/equ_${i}/4NPT -v
#删除中间文件
rm -f ./step1/equ_${i}/*.trr ./step1/equ_${i}/*.edr ./step1/equ_${i}/*tpr ./step1/equ_${i}/*.log ./step1/equ_${i}/*.xtc ./step1/equ_${i}/\#* > /dev/null 2>&1
done运行MD
经过上面的平衡后,接下来通过MD的配置文件执行MD。和上面的步骤一样,加权限后运行下面的脚本。 同样需要对副本温度进行修改。
#!/bin/bash
T="310.00, 310.79, 311.59, 312.38, 313.18, 313.98, 314.78, 315.58, 316.39, 317.19, 318.00, 318.81, 319.62, 320.44, 321.25, 322.07, 322.89, 323.70, 324.53, 325.35, 326.18, 327.00, 327.83, 328.66, 329.49, 330.33, 331.16, 332.00, 332.84, 333.68, 334.52, 335.37, 336.21, 337.06, 337.91, 338.76, 339.61, 340.47, 341.32, 342.18, 343.04, 343.90, 344.77, 345.63, 346.50, 347.37, 348.24, 349.12, 349.99, 350.87, 351.75, 352.63, 353.51, 354.39, 355.28, 356.17, 357.05, 357.95, 358.84, 359.73, 360.63, 361.53, 362.43, 363.33, 364.24, 365.16, 366.07, 366.98, 367.89, 368.80, 369.72, 370.64, 371.55, 372.48, 373.40, 374.33, 375.25, 376.18, 377.11, 378.05, 378.98, 379.92, 380.00"
arr_T=(${T//,/ }) #温度数据转为数组
len=${#arr_T[@]} #统计数组元素个数
mid=${len}-1
rm -r ./step2/MD* > /dev/null 2>&1 #删除step2路径下的残存文件夹
for ((i=0; i<=$mid; i++))
do
mkdir ./step2/MD_${i}
cp ./step2/mdp_file/* ./step2/MD_${i}/.
cp ./step1/equ_${i}/4*.gro ./step2/MD_${i}/.
sed -i 's/TEMP/'${arr_T[$i]}'/g' ./step2/MD_${i}/*.mdp
#成品模拟tpr文件生成
gmx_mpi grompp -f ./step2/MD_${i}/md.mdp -c ./step1/equ_${i}/4NPT.gro -p topol.top -o ./step2/MD_${i}/5MD.tpr
done通过下面命令运行MD
mpirun -np 8 gmx_mpi mdrun -deffnm 5MD -multidir ./step2/MD_[12345678] -replex 1000这里注意一下,-np代表调用的核心数,做副本交换分子动力学时,每个副本需要调用一个cpu,因此受限于cpu,如果cpu只有10个核心,那就只能运行十个副本。 运行完成后,通过下面的命令将轨迹整合,后面的分析和常规分子动力学模拟一样。
demux.pl ./step2/MD_1/5MD.log
gmx_mpi trjcat -f ./step2/MD_[12345678]/5MD.xtc -demux replica_index.xvg -o ./traj_merge/{1..8}_traj.xtc