基于 GROMACS 的小分子-蛋白复合物体系分子动力学模拟前准备
在分子动力学(MD)模拟中,准备工作是确保模拟结果可靠和精确的关键。特别是在模拟小分子-蛋白复合物体系时,涉及到分子拓扑、结构加氢以及力场的选择等多个环节。本篇文章将详细介绍如何为 GROMACS 分子动力学模拟提供配体(小分子)的拓扑信息,并探讨相关的底层技术。
1. 配体加氢处理
为什么需要加氢?
加氢是分子动力学模拟中的一项基本操作。在对接后生成的小分子往往缺乏氢原子,这会影响力场的精度和分子模拟的稳定性。通过对小分子进行加氢,我们能够确保其在模拟中具有准确的氢键结构,从而提高模拟的准确性和物理意义。
使用 Avogadro 进行加氢
Avogadro 是一种常用的分子可视化和编辑工具,能够方便地对分子进行加氢处理。加氢后的分子可以用于生成与 GROMACS 兼容的拓扑文件。
加氢步骤:
- 打开 Avogadro,导入对接得到的配体结构文件
out_ligand_1.pdb。 - 在顶部菜单中选择 Build → Add Hydrogens,Avogadro 会根据分子的化学环境自动添加氢原子。
- 添加完氢原子后,另存为
out_ligand_1_addH.pdb文件。
加氢后的分子会包含所有氢原子,确保了分子结构的完整性,适合后续的拓扑生成与动力学模拟。
2. 上传至 ATB 生成 ITP 文件
什么是 ITP 文件?
ITP(Individual Topology)文件是 GROMACS 用于描述小分子、离子、溶剂等的拓扑文件。它包含了分子的原子类型、质荷、电荷、键、角、二面角等力学参数。在小分子-蛋白复合物模拟中,ITP 文件提供了描述配体的必要信息。
ATB(Automatic Topology Builder)是一个在线工具,它能够根据上传的分子结构自动生成与 GROMACS 兼容的拓扑文件。ATB 会结合力场(如 GROMOS、AMBER 等)生成与分子结构相匹配的 ITP 文件。
上传和生成 ITP 文件的步骤:
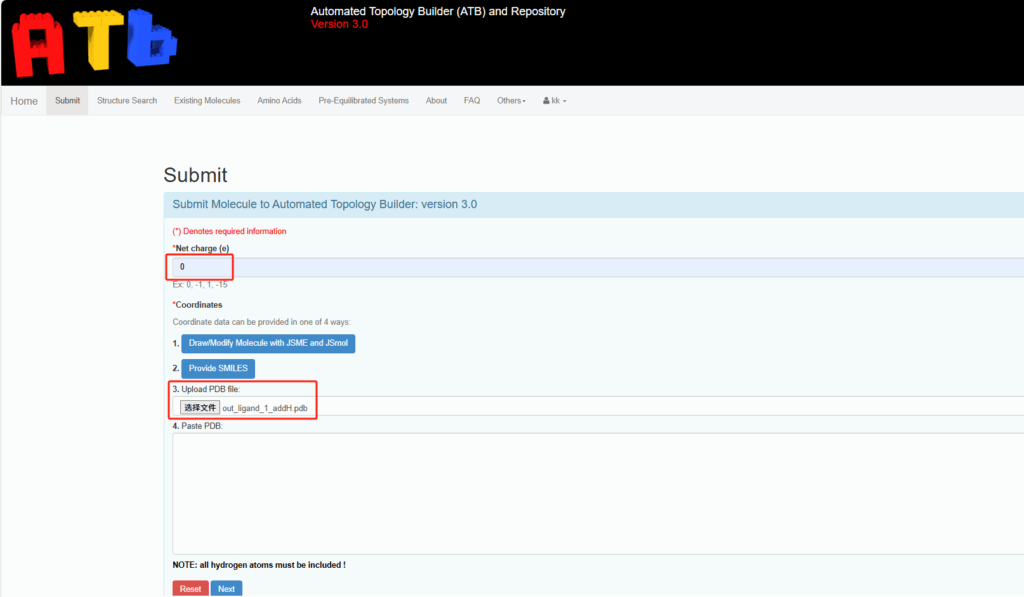
- 访问 ATB 网站 并登录。
- 将加氢后的分子结构文件
out_ligand_1_addH.pdb上传到 ATB 网站。 - 选择适当的力场(通常推荐 GROMOS 或 AMBER)。
- 点击生成 ITP 文件,ATB 会自动为配体生成与 GROMACS 兼容的拓扑文件。
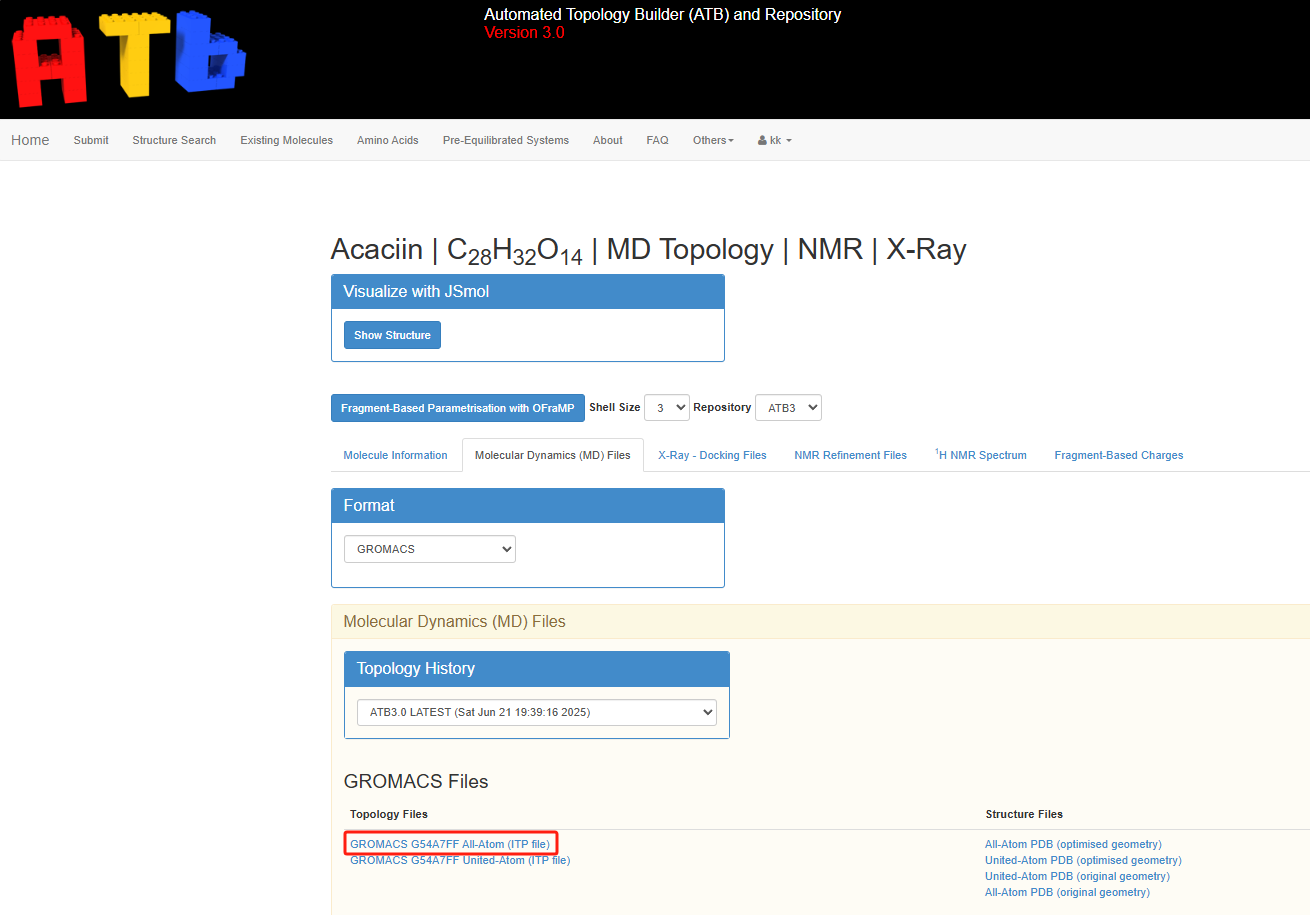
下载 ITP 文件
生成 ITP 文件后,可以在 ATB 的个人页面下找到对应的 Saved Molecules 栏目,点击 Molid ID,进入页面后会显示相应的 ITP 文件。右键点击链接并选择 另存为,保存为 LIG.itp 文件,或者复制文件内容并粘贴到本地文件 LIG.itp 中。
3. 准备其他必要的文件
在开始模拟之前,除了配体的拓扑文件外,还需要准备以下文件:
- LIG.itp:由 ATB 生成的小分子配体拓扑文件。
- out_ligand_1_addH.pdb:加氢后的配体结构文件。
- 蛋白.pdb:蛋白质的结构文件,通常可以从 PDB 数据库(Protein Data Bank)下载。
这些文件将作为输入,供 GROMACS 进行系统构建和模拟。
4. GROMACS 中的拓扑生成与力场基础
在 GROMACS 中,拓扑文件定义了分子系统中的原子、分子间相互作用以及力学模型。对于小分子-蛋白复合物系统,拓扑文件的构建通常包括以下内容:
- 原子类型与电荷:每个原子都需要定义其类型(如 C, N, O, H 等),并指定相应的电荷。
- 键、角、二面角:分子内的化学键、键角和二面角需要被定义,以计算分子间的力学相互作用。
- 非键相互作用:如范德华力、库伦力等,需要根据力场参数进行定义。
力场参数的应用
GROMACS 支持多种常用的力场(如 GROMOS、AMBER、CHARMM、OPLS 等)。这些力场通过一组预设的参数(如键长、键角、原子间的相互作用参数等)来描述分子体系的能量。以 GROMOS 力场为例,能量函数可以表示为:
$$ E = E{\text{bond}} + E{\text{angle}} + E{\text{dihedral}} + E{\text{nonbonded}} $$
其中:
-
( E{\text{bond}} ) 表示键能,通常使用简单的弹簧模型表示: $$ E{\text{bond}} = k_b (r – r_0)^2 $$ 其中 ( k_b ) 是键力常数,( r_0 ) 是平衡键长,( r ) 是当前键长。
-
( E{\text{angle}} ) 表示键角能,采用二次势能模型: $$ E{\text{angle}} = k_\theta (\theta – \theta_0)^2 $$
-
( E{\text{dihedral}} ) 是二面角能,通常使用周期性势能: $$ E{\text{dihedral}} = V_n \left[ 1 + \cos(n\phi – \gamma) \right] $$
-
( E{\text{nonbonded}} ) 是非键相互作用能,包括范德华力和库伦力: $$ E{\text{nonbonded}} = \sum{i
其中,( A ) 和 ( B ) 是范德华参数,( q_i ) 和 ( qj ) 是粒子的电荷,( r{ij} ) 是粒子间的距离。
这些参数通过力场定义,并在 GROMACS 模拟中被用来计算分子系统的能量和力。
准备其他文件
out_ligand_1_addH.pdb
蛋白.pdb
LIG.itp
立场文件
em.mdp
ions.mdp
nvt.mdp
npt.mdp
md.mdp执行命令
gmx editconf -f out_ligand_1_addH.pdb -o LIG.gro #将加氢的小分子转换成gro文件
gmx pdb2gmx -f 7alv-8etr.pdb -o receptor_processed.gro -water spce 选择力场54a7,选项1
# 构建complex.gro文件,将生成的蛋白gro文件receptor_processed.gro和小分子gro文件LIG.gro合并成complex.gro
# 构建Topology,在top文件中加入下面两行
; Include ligand topology
#include "LIG.itp"
# 在top文件最后一行加入LIG 1
[ molecules ]
; Compound #mols
Protein_chain_A 1
LIG 1
# 将复合物体系置于周期性水盒子中
gmx editconf -f complex.gro -o newbox.gro -bt dodecahedron -d 1.0
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
# 平衡电荷,加钠是np,加氯是nn
gmx grompp -f ions.mdp -c solv.gro -p topol.top -o ions.tpr -maxwarn 200
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -np 6
# 能量最小化
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr -maxwarn 200
gmx mdrun -v -deffnm em -nb gpu
# 施加约束力
gmx genrestr -f LIG.gro -o posre_LIG.itp -fc 1000 1000 1000
# 修改top文件
; Ligand position restraints
#ifdef POSRES
#include "posre_LIG.itp"
#endif
gmx make_ndx -f em.gro -o index.ndx
> 1 | 13
> q
# nvt系综平衡模拟
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr -maxwarn 200
gmx mdrun -v -deffnm nvt -nb gpu
# npt系综平衡模拟
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr -maxwarn 200
gmx mdrun -deffnm npt -v -nb gpu
# 运行md
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_1.tpr -maxwarn 200
gmx mdrun -deffnm md_0_1 -v -nb gpu